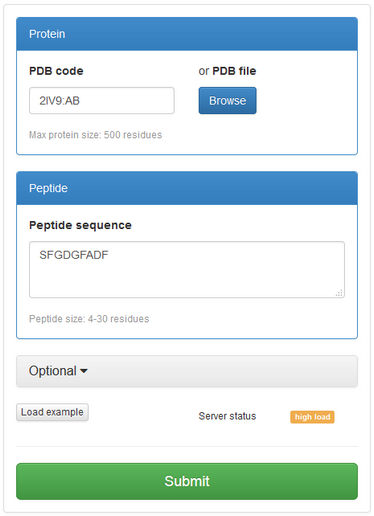
See an example screenshot of the “Submit new job” form on the right:
The only required data is protein structure and peptide sequence.
Protein receptor structure can be provided as file in the pdb format or protein pdb code along with the chain identifier(s), for example: 1RJK:A or 1CE1:HL (the structure is automatically obtained from Protein Data Bank database).
Peptide sequence should be entered in single-letter amino acid code. Allowed size of the peptide is 3-30 amino acids.
After providing protein structure and peptide sequence, you can simply run simulation by clicking “Submit” button. However, it is possible to set some more parameters in “Optional” tab (see below for more details).
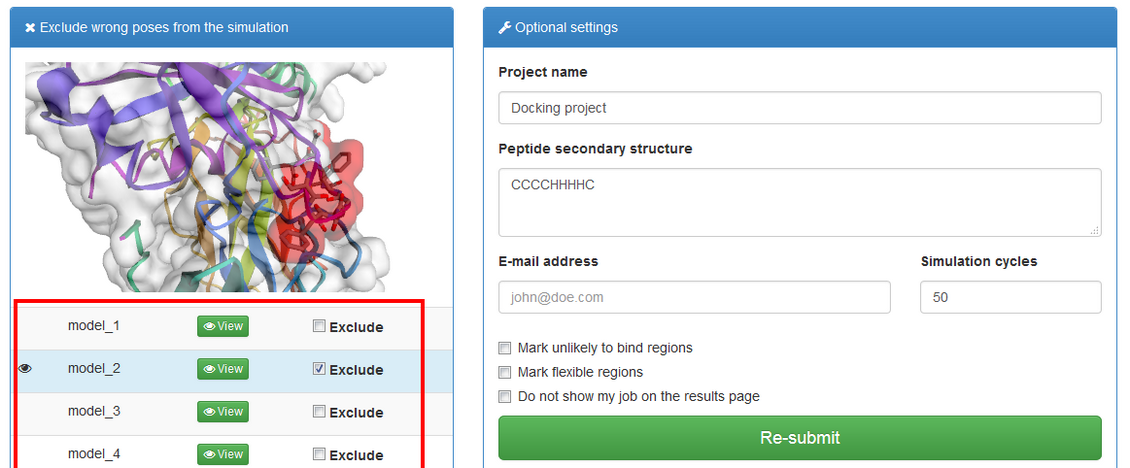
In “optional” settings tab you can:
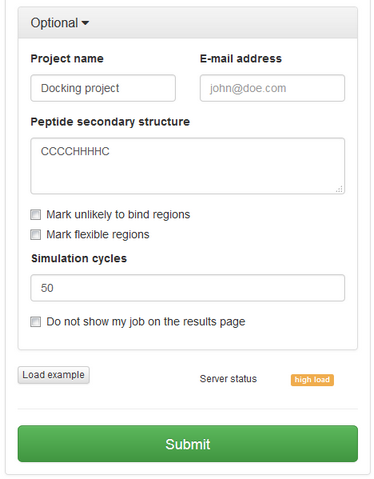
See an example
If “Mark flexible regions” is checked, after clicking “submit button” you will be redirected to the panel shown below:
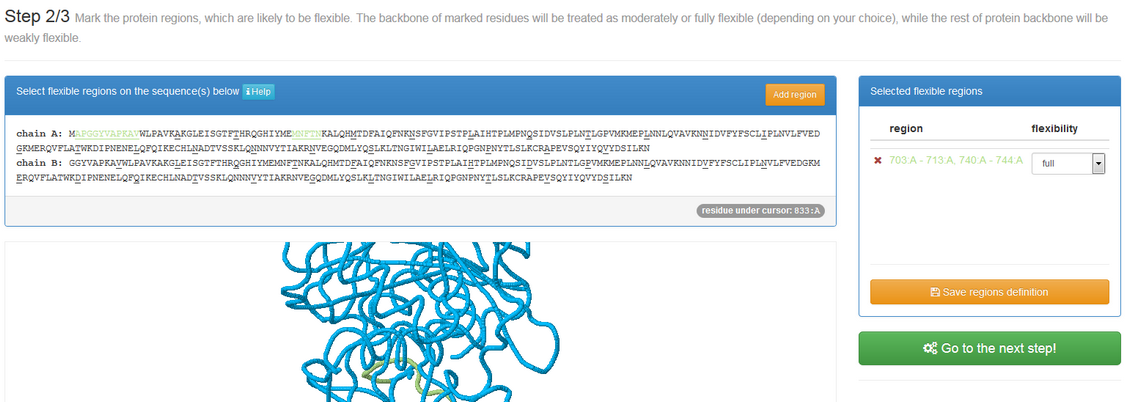
See an example
If “Mark unlikely to bind regions” is checked you will be redirected to the panel which is similar to one described in the previous subsection:
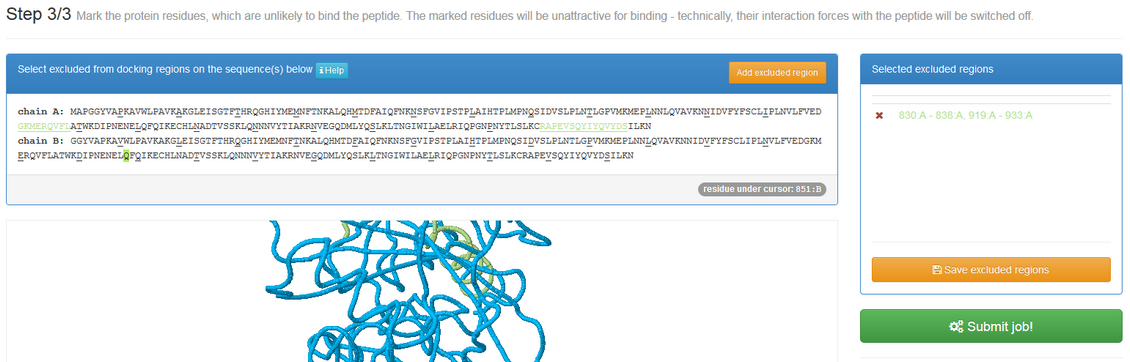
Here you can select receptor residues that are unlikely to interact with the peptide to exclude some binding modes from the results.
1060:C 6:PEP 5.0 1.01060:C 6:PEP 5.0 1.01066:C 7:PEP 5.0 1.01060:C 6:PEP 5.0 1.01060:C 6:PEP 5.01060:C 6:PEP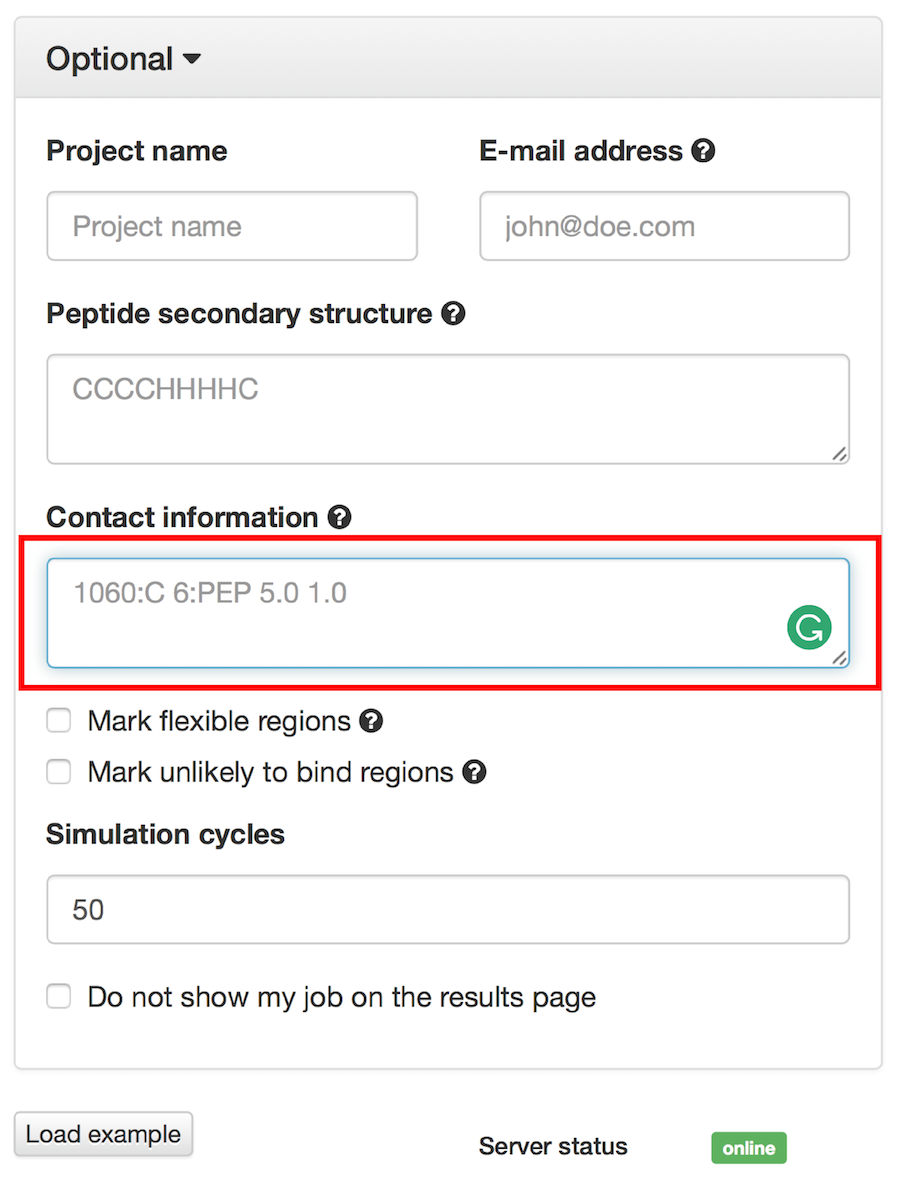
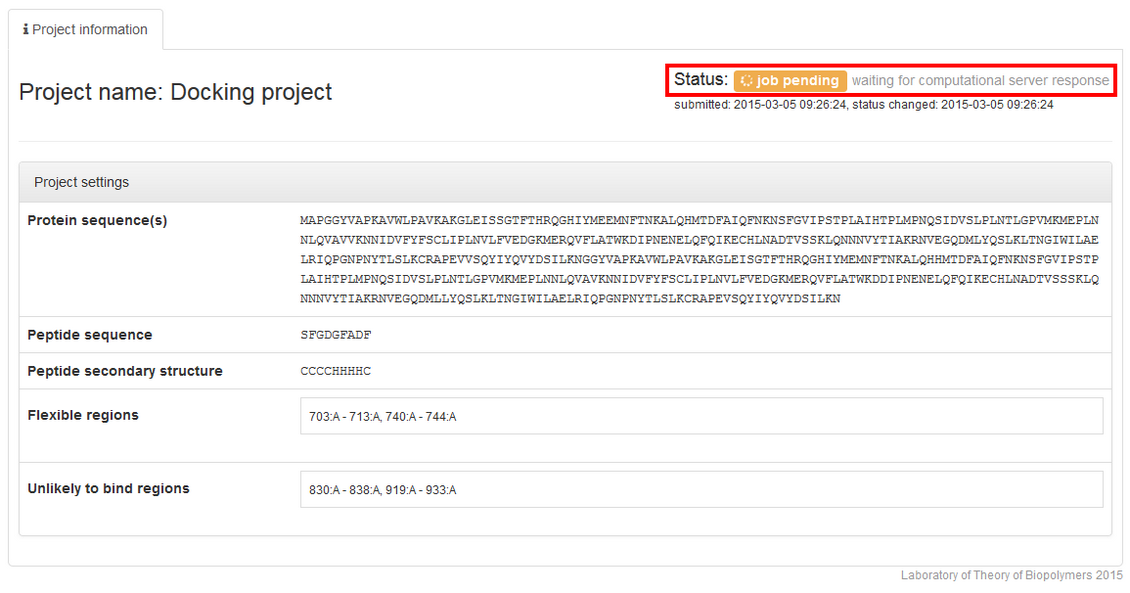
After clicking the Submit button, you will be redirected to project information page.
The job status information will start from the job pending status:
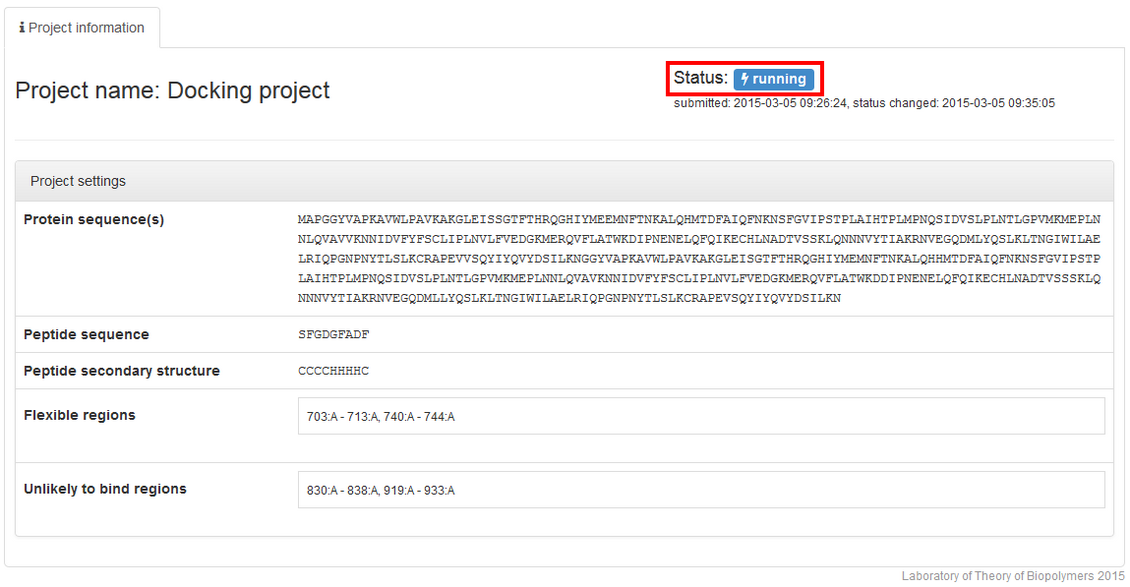
followed by the running status:
The job ends with the done status, and additional tabs containing results are provided:
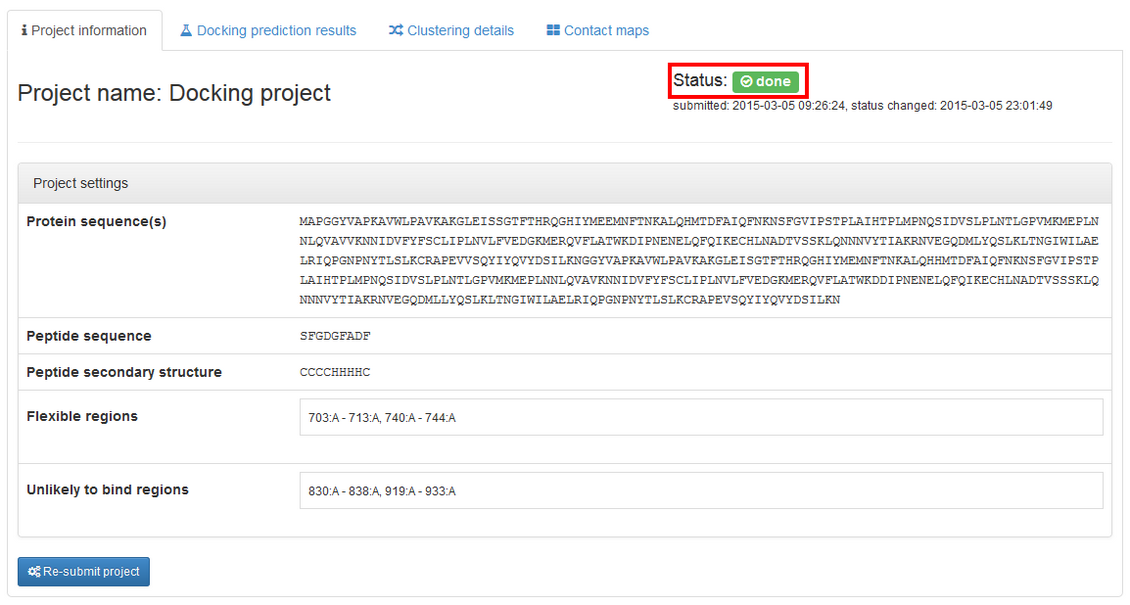
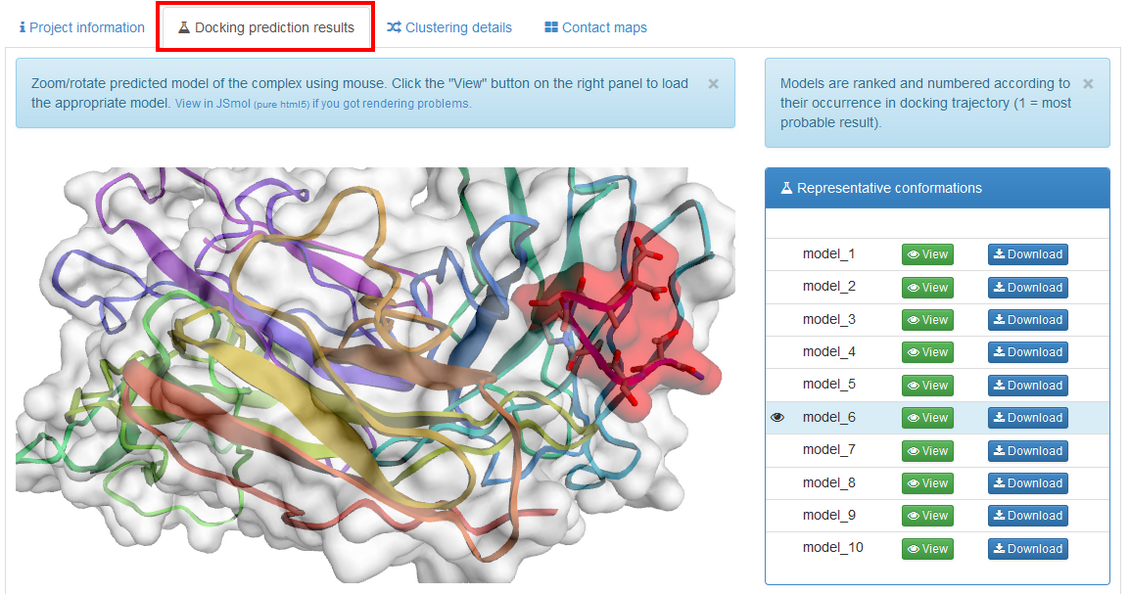
The left panel of the "Docking prediction results" contains visualization of the predicted complex (the peptide is shown in red). You can easily rotate and zoom the model using mouse. If the model is not displayed, please check if Web Graphics Library (webGL) is enabled in your browser (see the WebGL test page for more details). Alternatively, you can switch to JSmol which is an HTML5 JavaScript-only web app.
The right panel contains list of the resulting models. They are derived from the modeling trajectory by means of hierarchical clustering method – all models from the trajectory are divided into 10 groups (clusters) of similar structures. For each of the cluster one representative model is selected. Models are then ranked according to density of the clusters (model_1 represents the most dense cluster) and reconstructed to all-atom representation.
model_1 (from the most dense cluster) is shown. To view other models click "View" button. You can also download models as pdb files by clicking "Download".
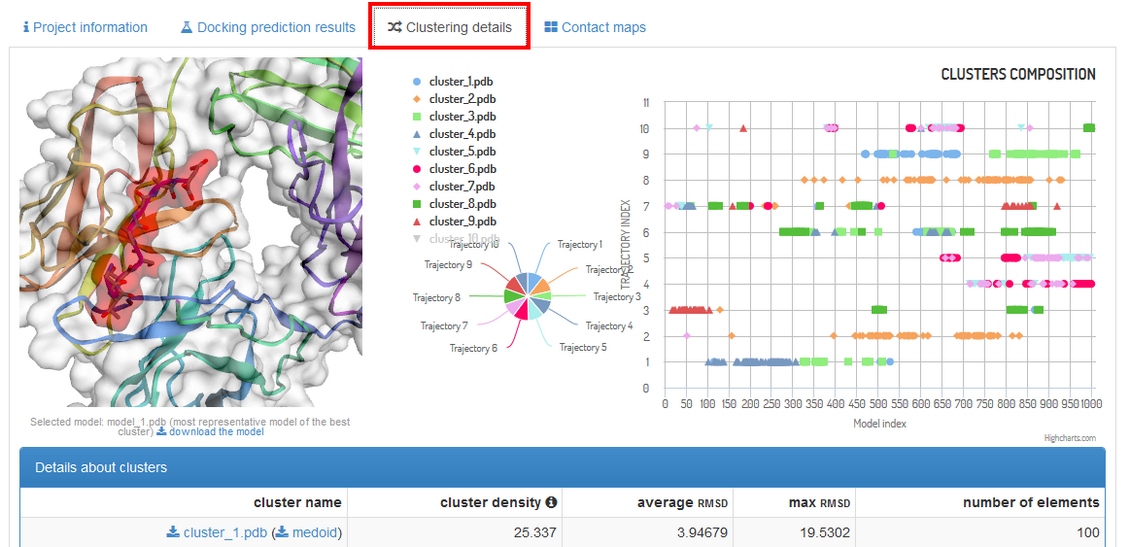
Under the “Clustering details” tab (see the screenshot below) you are provided with
thorough insight into structural clustering data.
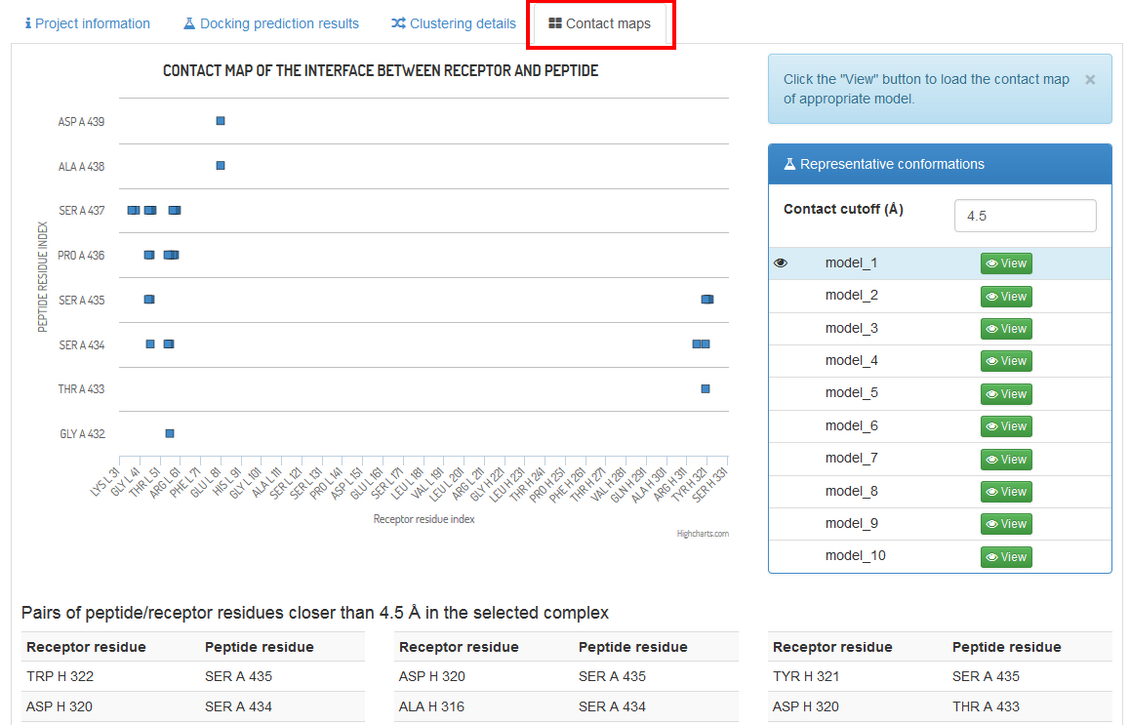
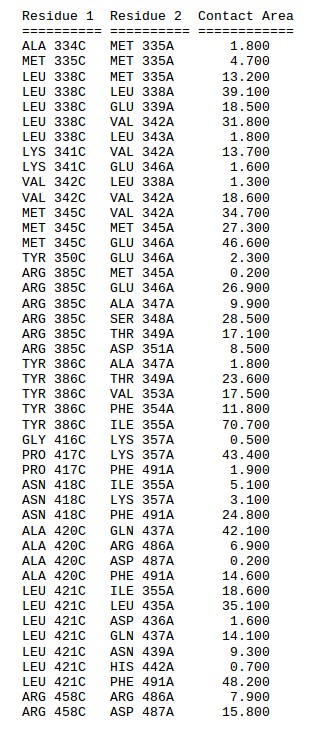
The “Contact maps” tab allows to investigate the interaction interface between the peptide and the receptor. An interactive chart shows a contact map between the peptide and the receptor residues. You may define the value of the contact cutoff distance i.e. distance between the closest heavy atoms of the residues (default value is 4.5 Å). At the bottom of the page a text list of the contacts shown on the maps.
From “Docking predictions results” tab, you may download a compressed archive with the output data. The archive file contains the following data:
model_*.pdb - 10 final models (in the PDB format and all-atom representation)cluster_*.pdb - cluster models (groups of models that have been classified in structural clustering to particular clusters; in the PDB format and C-alpha representation)
Cluster numbering correspond to models numbering i.e. model_7.pdb is a representative model of models grouped in the seventh (ranked as seventh) cluster (cluster_7.pdb).trajectory_*.pdb - complete trajectories (in the PDB format and C-alpha representation)top1000.pdb - top 1000 models (in the PDB format and C-alpha representation)input.pdb - the input structure of the receptor
README - a log file with all information to recreate the simulationenergy.txt - log file from the simulation with energy and its distribution; the
columns contain the following data:
For 10 final models, the CABS-dock server provides automated reconstruction to all-atom representation and local optimization using Modeller procedure (the optimization is done by the minimization with the DOPE, Discreet Optimized Protein Energy, statistical potential). Larger sets of CABS-dock models (1000 or 10,000, downloaded from the server) can be reconstructed by a user using standalone Modeller package.
The script for the Modeller reconstruction and optimization procedure is available here
.
All downloadable pdb files (models, clusters, and trajectories available from the ‘Docking predictions results’ tab) can be easily viewed using molecular visualization software, like Pymol.
Clusters and trajectories contains only Cα atoms.
In order to view Cα trace load the pdb file into Pymol and type:
show ribbon set ribbon_trace_atoms, 1
show ribbon set ribbon_trace_atoms, 1 mplayIf you want to view all models at ones type:
set all_states, 1
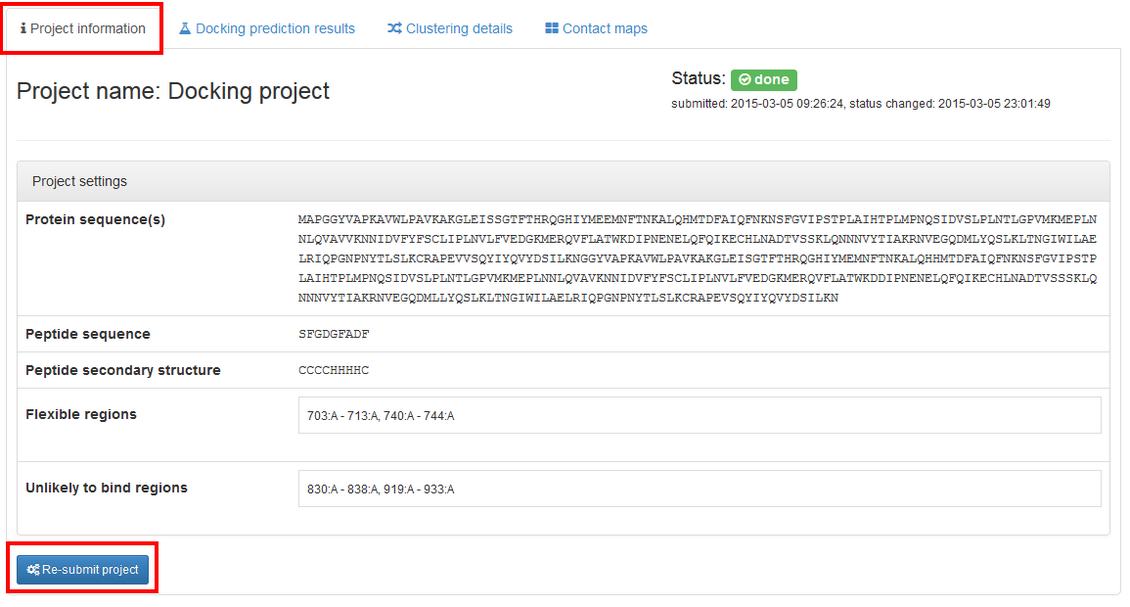
You may resubmit your job with changed job parameters. To do so go to the “Project information” tab and press Re-submit project.
An example result page can be accessed here. See also an "Examples" section
CABS-dock webserver may be operated throught RESTful services, using following URIs:
https://biocomp.chem.uw.edu.pl/CABSdock/REST/status/somejobidentifier check job status onlyhttps://biocomp.chem.uw.edu.pl/CABSdock/REST/job_info/somejobidentifier check information about
submitted job
https://biocomp.chem.uw.edu.pl/CABSdock/REST/add_job/ submit new job
receptor_pdb_code PDB code of receptor with or without chain inforeceptor_file Send your PDB file with receptor
ligand_seq Ligand sequenceproject_name project nameemail email (info about job will be send to this email)ligand_ss Ligand secondary structuresimulation_cycles Number of simulation cycles (max 200)show_job ("True" or "False") if False - do not show job on the
queue page
excluded_regions Array of excluded regions. You can select receptor residues that are
unlikely to interact with the peptide.(start position, end position, chain)
flexible_regions Array of flexible regions (removing distance restraints that keep the receptor
structure in a near native conformation). (start position, end position, chain, flexibility (full / moderate))
https://biocomp.chem.uw.edu.pl/CABSdock/REST/job_results_all/somejobidentifier Get full results for
job. Optional filter for attribute (min/max/range)
value Attributemin Minimal valuemax Maximum valuedensitycluster densityrmsd Average RMSDmaxrmsd Max RMSDcounts Number of elementshttps://biocomp.chem.uw.edu.pl/CABSdock/REST/job_results/somejobidentifier Get results for job
(most interesting informations). Optional filter for attribute (min/max/range)
value Attributemin Minimal valuemax Maximum valuedensitycluster densityrmsd Average RMSDmaxrmsd Max RMSDcounts Number of elementshttps://biocomp.chem.uw.edu.pl/CABSdock/REST/get_cluster/somejobidentifier/cluster_number Get
cluster results.
https://biocomp.chem.uw.edu.pl/CABSdock/REST/get_trajectory/somejobidentifier/trajectory_number Get
trajectory.
https://biocomp.chem.uw.edu.pl/CABSdock/REST/get_selected_trajectory/somejobidentifier/cluster_number/start/end
Get selected part of trajectory. Start and end must be in the range (1,1000)
Submit job for XYZ protein and SFDG ligand sequence, using cURL, with default parameters :
curl -H "Content-Type: application/json" -X POST -d '{"receptor_pdb_code":"XYZ", "ligand_seq":"SFGD"}'
https://biocomp.chem.uw.edu.pl/CABSdock/REST/add_job/.
User should get output like:
HTTP/1.0 200 OK
Content-Type: application/json
Content-Length: 30
Server: Werkzeug/0.10.4 Python/2.7.6
Date: Wed, 20 Jul 2015 19:53:23 GMT
{
"jid": "a6433216640dcb3"
}
where jid is job identifier assigned to the just submitted job. Otherwise (for example if pdbcode
doesn't exists or input data doesn't fullfill requirements), user should get output similar to:
HTTP/1.0 404 NOT FOUND Content-Type: text/html; charset=utf-8 Content-Length: 5521 Server: Werkzeug/0.9.6 Python/2.7.8 Date: Wed, 12 Nov 2014 13:56:48 GMT [...]or raise error
HTTP/1.0 200 OK
Content-Type: application/json
Content-Length: 165
Server: Werkzeug/0.10.4 Python/2.7.6
Date: Wed, 22 Jul 2015 20:02:41 GMT
{
"error": "Protein code must be 4-letter (2PCY) or >5 letters (2PCY:A or 2PCY:AB ...)"
}
To override default parameters, user may post additional options, i.e.:
curl -H "Content-Type: application/json" -X POST -d '{"receptor_pdb_code":"2IV9",
"ligand_seq":"SFGD","project_name":"my_project1", "email":"mail@host.com", "ligand_ss":"CCHHC",
"simulation_cycles":"100", "show_job":True, "excluded_regions":[{"start":"100","end":"340","chain":"A"}],
"flexible_regions":[{"start":"101","end":"202","chain":"B","flexibility":"full"}]}'
https://biocomp.chem.uw.edu.pl/CABSdock/REST/add_job/
import requests
import json
url = 'https://biocomp.chem.uw.edu.pl/CABSdock/REST/add_job/'
files = {'file': open('your_PDB_file.pdb')} #or use PDB code in var data
data = {
"receptor_pdb_code": "2IV9", #or use PDB file in var files
"ligand_seq": "SFGD",
"email": "mail@host.com",
"show": True,
"project_name":"my_project1",
"excluded_regions":[
{
"start": "1000",
"end": "2000",
"chain": "A"
}
],
"flexible_regions":[
{
"start": "101",
"end": "202",
"chain": "A",
"flexibility": "full"
},
{
"start": "300",
"end": "370",
"chain": "B",
"flexibility": "moderate"
},
]
}
response = requests.post(url, files=files, data=data) #request with file
#response = requests.post(url, data=data) # request without file
print response.text
HTTP/1.0 200 OK
Content-Type: application/json
Content-Length: 22
Server: Werkzeug/0.10.4 Python/2.7.6
Date: Sun, 26 Jul 2015 10:31:35 GMT
{
"status": "done"
}
done Job is done - results are readypending / running / pre_quere Job is in progresserror Job ID is faultyimport requests import json url = 'https://biocomp.chem.uw.edu.pl/CABSdock/REST/status/somejobidentifier' response = requests.get(url) print response.text
HTTP/1.0 200 OK
Content-Type: application/json
Content-Length: 929
Server: Werkzeug/0.10.4 Python/2.7.6
Date: Sun, 26 Jul 2015 11:06:21 GMT
{
"del": "Mon, 26 Oct 2015 19:11:32 GMT",
"excluded": [
"783:A - 937:A"
],
"flexible": [
"767:A - 937:A"
],
"ligand_chain": "A",
"ligand_sequence": "SFGDGFADF",
"ligand_ss": "CCCCHHHHC",
"project_name": "Docking project",
"receptor_sequence": "MAPGGYVAPKAVWLPAVKAKGLEISSGTFTHRQGHIYMEEMNFTNKALQHMTDFAIQFNKNSFGVIPSTPLAIHTPLMPNQSIDVSLPLNTLGPVMKMEPLNNLQVAVV",
"ss_psipred": 0,
"status": "pending",
"status_change": "Sun, 28 Jun 2015 19:11:32 GMT",
"status_date": "Sun, 28 Jun 2015 19:11:32 GMT"
}
import requests import json url = 'https://biocomp.chem.uw.edu.pl/CABSdock/REST/job_info/somejobidentifier' response = requests.get(url) print response.text
To get result send
curl -i https://biocomp.chem.uw.edu.pl/CABSdock/REST/job_results/somejobidentifier
We strongly recommend to send curl with compression
curl -i -H 'Accept-Encoding: gzip,deflate'
https://biocomp.chem.uw.edu.pl/CABSdock/REST/job_results/somejobidentifier
{
"models": [
{
"average_rmsd": "5.29163",
"cluster_density": "18.8978",
"elements": "100",
"info": {
"del": "Wed, 07 Oct 2015 21:34:27 GMT",
"ligand_chain": "A",
"ligand_sequence": "MGTEATEQVSWGHYSGDEEDAYSAEPLPEL",
"ligand_ss": "CCCCCCCCCCCCCCCCCCCCCCCCCCCCCC",
"project_name": "87aeb5bf523d141",
"receptor_sequence": "AILPIASSCCTEVSHHISRRLLERVNMCRIQRADGDCDLAAVILHVKRRRICVSPHNHTVKQWMKVQAAKKNGKGNVCHRKKHHGK",
"ss_psipred": 1,
"status": "done",
"status_change": "Tue, 09 Jun 2015 23:00:30 GMT",
"status_date": "Tue, 09 Jun 2015 21:34:27 GMT"
},
"jobid": "87aeb5bf523d141",
"max_rmsd": "28.8464",
"model": 1,
"model_data": "REMARK 1 secondary structure assigned by stride \nREMARK 2 stride output converted by stride2pdb version 0.01\nHELIX 1 1 ARG X 19 GLU X 23 1 5\nHELIX 2 2 HIS X 58 ALA X 69 1 [[....................]]"
},
{
"average_rmsd": "7.75161",
"cluster_density": "16.5127",
"elements": "128",
"info": {
"del": "Wed, 07 Oct 2015 21:34:27 GMT",
"ligand_chain": "A",
"ligand_sequence": "MGTEATEQVSWGHYSGDEEDAYSAEPLPEL",
"ligand_ss": "CCCCCCCCCCCCCCCCCCCCCCCCCCCCCC",
"project_name": "87aeb5bf523d141",
"receptor_sequence": "AILPIASSCCTEVSHHISRRLLERVNMCRIQRADGDCDLAAVILHVKRRRICVSPHNHTVKQWMKVQAAKKNGKGNVCHRKKHHGK",
"ss_psipred": 1,
"status": "done",
"status_change": "Tue, 09 Jun 2015 23:00:30 GMT",
"status_date": "Tue, 09 Jun 2015 21:34:27 GMT"
},
"jobid": "87aeb5bf523d141",
"max_rmsd": "28.2751",
"model": 2,
"model_data": "REMARK 1 secondary structure assigned by stride \nREMARK 2 stride output converted by stride2pdb version 0.01\nHELIX 1 1 ARG X 19 LEU X 22 1 4\nHELIX 2 2 THR X 59 ALA X 69 1 11\nSHEET 1 1 1 MET X 27 CYS X 28 0\nSHEET 2 2 1 LEU X 44 HIS X 45 0\nSHE [[....................]] "
},
{},{},{},....,{}
}
To get filtered results
curl -i -X POST -d '{"filter":"density","min":"10","max":"20"}'
https://biocomp.chem.uw.edu.pl/CABSdock/REST/job_results/somejobidentifier
import requests
import json
url = 'https://biocomp.chem.uw.edu.pl/CABSdock/REST/job_results/somejobidentifier'
#filtering is optional
data = {
"value":"rmsd",
"min":"5",
"max":"12"
}
response = requests.post(url,data=data)
print response.text
To get result send
curl -i https://biocomp.chem.uw.edu.pl/CABSdock/REST/job_results_all/somejobidentifier
We strongly recommend to send curl with compression
curl -i -H 'Accept-Encoding: gzip,deflate'
https://biocomp.chem.uw.edu.pl/CABSdock/REST/job_results_all/somejobidentifier
{
"models": [
{
"average_rmsd": "5.29163",
"cluster_data": "MODEL 102\nATOM 1 CA ALA X 3 -0.304 -0.446 -12.449 1.00 1.00 C\nATOM 2 CA ILE X 4 2.456 1.179 -13.176 3.00 1.00 C\nATOM 3 [.....]",
"cluster_density": "18.8978",
"elements": "100",
"info": {
"del": "Wed, 07 Oct 2015 21:34:27 GMT",
"ligand_chain": "A",
"ligand_sequence": "MGTEATEQVSWGHYSGDEEDAYSAEPLPEL",
"ligand_ss": "CCCCCCCCCCCCCCCCCCCCCCCCCCCCCC",
"project_name": "87aeb5bf523d141",
"receptor_sequence": "AILPIASSCCTEVSHHISRRLLERVNMCRIQRADGDCDLAAVILHVKRRRICVSPHNHTVKQWMKVQAAKKNGKGNVCHRKKHHGK",
"ss_psipred": 1,
"status": "done",
"status_change": "Tue, 09 Jun 2015 23:00:30 GMT",
"status_date": "Tue, 09 Jun 2015 21:34:27 GMT"
},
"jobid": "87aeb5bf523d141",
"max_rmsd": "28.8464",
"model": 1,
"model_data": "REMARK 1 secondary structure assigned by stride \nREMARK 2 stride output converted by stride2pdb version 0.01\nHELIX 1 1 ARG X 19 GLU X 23 1",
"trajectory_data": "MODEL 1\nREMARK CABSDock Temp 6.45\nREMARK CABSDock Erec -29.49\nREMARK CABSDock Elig -29.39\nREMARK CABSDock Eint 0.00\nREMARK CABSDock Etot -58.88\nATOM 1 CA ALA X 3 -1.057"
},
{},{},{},....,{}
}
To get filtered results
curl -i -X POST -d '{"filter":"rmsd","min":"5","max":"12"}'
https://biocomp.chem.uw.edu.pl/CABSdock/REST/job_results_all/somejobidentifier
import requests
import json
url = 'https://biocomp.chem.uw.edu.pl/CABSdock/REST/job_results_all/somejobidentifier'
#filtering is optional
data = {
"value":"rmsd",
"min":"5",
"max":"12"
}
response = requests.get(url,data=data)
print response.text
To get cluster send
curl -i
https://biocomp.chem.uw.edu.pl/CABSdock/REST/get_cluster/somejobidentifier/clusterNumber
where clusterNumber
is number between [1,10] corresponding to model number.
We strongly recommend to send curl with compression
curl -i -H 'Accept-Encoding:
gzip,deflate' https://biocomp.chem.uw.edu.pl/CABSdock/REST/get_cluster/somejobidentifier/clusterNumber
{
"model": [
{
"average_rmsd": "8.45027",
"cluster_data": "MODEL 101\nATOM 1 CA LEU A 6 28.654 -0.817 25.718 1.00 1.00 C\nATOM 2 CA GLY A 7 31.720 -3.251 26.268 3.00 1.00 [.......]",
"cluster_density": "29.3482",
"elements": "248",
"jobid": "8ae372b8e877be4",
"max_rmsd": "41.6541"
}
]
}
To get trajectory send
curl -i
https://biocomp.chem.uw.edu.pl/CABSdock/REST/get_trajectory/somejobidentifier/modelNumber
where modelNumber
is between [1,10]
We strongly recommend to send curl with compression
curl -i -H 'Accept-Encoding: gzip,deflate'
https://biocomp.chem.uw.edu.pl/CABSdock/REST/get_trajectory/somejobidentifier/modelNumber
{
"model": [
{
"jobid": "8ae372b8e877be4",
"trajectory": "MODEL 1\nREMARK CABSDock Temp 6.45\nREMARK CABSDock Erec -1529.95\nREMARK CABSDock Elig -7.95\nREMARK CABSDock Eint 0.00\nREMARK CABSDock Etot -1537.90\nATOM 1 CA LEU A 6 [.........]"
}
]
}
Additionally, you can take a section of the trajectory model
curl -i
https://biocomp.chem.uw.edu.pl/CABSdock/REST/get_selected_trajectory/somejobidentifier/cluster_number/start/end
The example with the default CABS-dock server settings using the following data:
import requests
import json
url = 'http://0.0.0.0:5000/REST/add_job/'
files = {'file': open('your_PDB_file.pdb')} #or use PDB code in var data
data = {
"receptor_pdb_code": "2AM9"
"ligand_seq": "SSRFESLFAG",
"ligand_ss": "CHHHHHHHHC"
response = requests.post(url, data=data)
print response.text
}
response = requests.post(url, files=files, data=data) #request with file
#response = requests.post(url, data=data) # request without file
print response.text
For each selected residue, the user may choose from two preset settings: moderate or full flexibility. Technically this is achieved by changing the default distance restrains (used to keep the receptor structure near to the input conformation). The assignment of moderate flexibility decreases the strength of restrains, while full flexibility assigned removes all the restraints imposed on the selected residue.
Example:
import requests
import json
url = 'http://0.0.0.0:5000/REST/add_job/'
files = {'file': open('your_PDB_file.pdb')} #or use PDB code in var data
data = {
"receptor_pdb_code": "2RTM"
"ligand_seq": "HPQFEK",
"ligand_ss": "CHHHCC"
"flexible_regions":[
{
"start": "45",
"end": "54",
"chain": "A",
"flexibility": "full"
}
]
}
response = requests.post(url, data=data)
print response.text
}
response = requests.post(url, files=files, data=data) #request with file
#response = requests.post(url, data=data) # request without file
print response.text
In the default mode, CABS-dock allows peptides to explore the entire receptor surface. However, in certain modeling cases it is known that some parts of the protein are not accessible (for example due to binding to other proteins) and therefore it could be useful to exclude these regions from the search procedure.
Example:
import requests
import json
url = 'http://0.0.0.0:5000/REST/add_job/'
files = {'file': open('your_PDB_file.pdb')} #or use PDB code in var data
data = {
"receptor_pdb_code": "1CZY:C"
"ligand_seq": "PQQATDD",
"ligand_ss": "CEECCCC"
"excluded_regions":[
{
"start": "334",
"end": "335",
"chain": "C",
},
{
"start": "338",
"end": "338",
"chain": "C",
},
{
"start": "341",
"end": "342",
"chain": "C",
},
{
"start": "345",
"end": "345",
"chain": "C",
},
{
"start": "350",
"end": "350",
"chain": "C",
},
{
"start": "385",
"end": "386",
"chain": "C",
},
{
"start": "416",
"end": "418",
"chain": "C",
},
{
"start": "420",
"end": "421",
"chain": "C",
},
{
"start": "458",
"end": "458",
"chain": "C",
}
]
}
response = requests.post(url, data=data)
print response.text