
Source:
Journal of Biomolecular Structure and Dynamics, 16:381–396, 1998Abstract
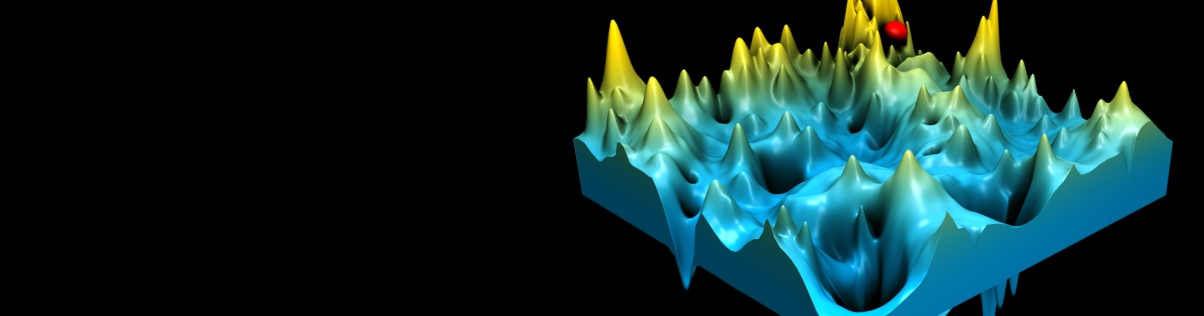
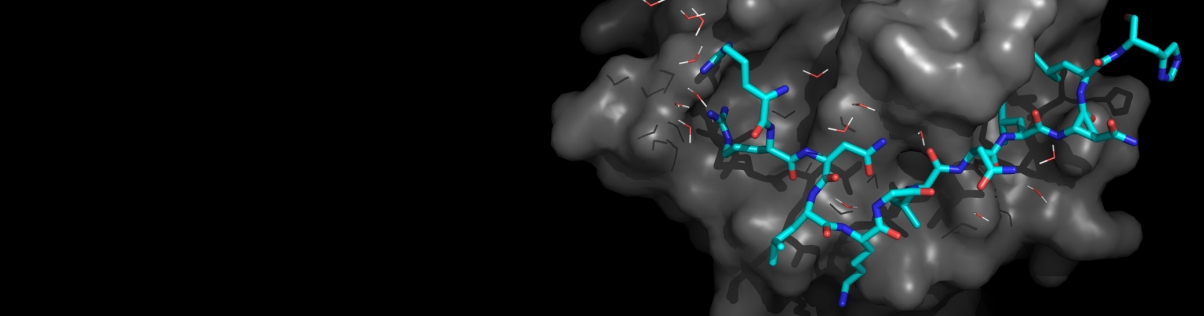
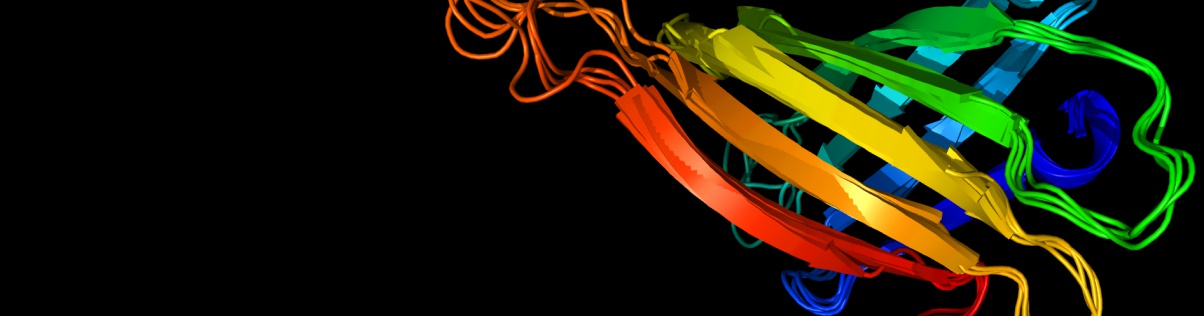
One of the most important unsolved problems of computational biology is prediction of the three-dimensional structure of a protein from its amino acid sequence. In practice, the solution to the protein folding problem demands that two interrelated problems be simultaneously addressed. Potentials that recognize the native state from the myriad of misfolded conformations are required, and the multiple minima conformational search problem must be solved. A means of partly surmounting both problems is to use reduced protein models and knowledge-based potentials. Such models have been employed to elucidate a number of general features of protein folding, including the nature of the energy landscape, the factors responsible for the uniqueness of the native state and the origin of the two-state thermodynamic behavior of globular proteins. Reduced models have also been used to predict protein tertiary and quaternary structure. When combined with a limited amount of experimental information about secondary and tertiary structure, molecules of substantial complexity can be assembled. If predicted secondary structure and tertiary restraints are employed, low resolution models of single domain proteins can be successfully predicted. Thus, simplified protein models have played an important role in furthering the understanding of the physical properties of proteins.