pyCABS API¶
pyCABS Copyright (C) 2013 Michal Jamroz <jamroz@chem.uw.edu.pl>
This program is free software: you can redistribute it and/or modify it under the terms of the GNU General Public License as published by the Free Software Foundation, either version 3 of the License, or (at your option) any later version.
This program is distributed in the hope that it will be useful, but WITHOUT ANY WARRANTY; without even the implied warranty of MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the GNU General Public License for more details.
You should have received a copy of the GNU General Public License along with this program. If not, see <http://www.gnu.org/licenses/>.
- class pycabs.CABS(sequence, secondary_structure, templates_filenames, project_name)¶
CABS main class.
Warning
Manually update self.FF variable here (path to the FF directory with CABS files)
Parameters: - sequence (string) – one line sequence of the target protein
- secondary_structure (string) – one line secondary structure for the target protein
- templates_filenames (list) – path to 3D protein model templates in pdb file format which you want to use for modeling. Cα numbering in templates must be aligned to target sequence
- project_name (string) – project_name and working directory name (uniq)
- convertPdbToDcd(catdcd_path='/home/user/pycabs/FF/catdcd')¶
This is only simple wrapper to CatDCD software (http://www.ks.uiuc.edu/Development/MDTools/catdcd/), could be usable since *.dcd binary format is few times lighter than pdb, and many python libraries (ProDy, MDAnalysis) use *.dcd as trajectory input format. Before use, download CatDCD from http://www.ks.uiuc.edu/Development/MDTools/catdcd/ and modify catdcd_path.
- createLatticeReplicas(start_structures_fn=[], replicas=20)¶
Create protein models projected onto CABS lattice, which will be used as replicas.
Parameters: - start_structures_fn (list) – list of paths to pdb files which should be used instead of templates models. This parameter is optional, and probably not often used. Without it script creates replicas from templates files.
- replicas (integer) – define number of replicas in CABS simulation. However 20 is optimal for most cases, and you don’t need to change it in protein modeling case.
Note
If number of replicas is smaller than number of templates - program will create replicas using first replicas templates. If there is less templates than replicas, they are creating sequentially using template models.
- generateConstraints(exclude_residues=[], other_constraints=[])¶
Calculate distance constraints using templates 3D models. Constraint will be a square well of size d-std_dev-1.0,d+std_dev+1.0, where d is mean distance among templates between Cα atoms (if constraint will be exceeded, there is penalty, scaled by weight.
Weight is defined as a fraction of particular average distance among templates i.e. if pair of residues exist in 2 of 3 templates, weight will be 0.66. Using multiple sequence alignments it should provide stronger constraints on consistently aligned parts.
Parameters: - exclude_residues (list) – indexes of residues without constrains
- constrains (other) – user-defined constrains as list of tuples: (residue_i_index,residue_j_index,distance, constraint_strength)
- generateConstraintsOld(exclude_residues=[], other_constraints=[])¶
Calculate distance constraints using templates 3D models. Constraint will be a square well of size min(d), max(d) where d is mean distance among templates between Cα atoms (if constraint will be exceeded, there is penalty, scaled by weight.
Weight is defined as a fraction of particular average distance among templates i.e. if pair of residues exist in 2 of 3 templates, weight will be 0.66. Using multiple sequence alignments it should provide stronger constraints on consistently aligned parts.
Parameters: - exclude_residues (list) – indexes of residues without constrains
- constrains (other) – user-defined constrains as list of tuples: (residue_i_index,residue_j_index,distance, constraint_strength)
- getEnergy()¶
Read CABS energy values into list
Returns: list of models energy
- getTraCoordinates()¶
Read trajectory file into 2D list of coordinates
Returns: 2D list of trajectory coordinates ( list[1][6] is sixth coordinate of second trajectory model = z coordinate of second atom of second model)
- loadSGCoordinates()¶
Read center of mass of sidegroups from TRASG file
Returns: 2D list of sidechains coordinates
- modeling(Ltemp=1.0, Htemp=2.0, cycles=100, phot=300, constraints_force=1.0, dynamics=False)¶
Start CABS modeling
param Ltemp: Low temperature for Replica Exchange Monte Carlo type Ltemp: float param Htemp: High temperature for Replica Exchange Monte Carlo type Htemp: float param cycles: number of Replica Exchange cycles type cycles: integer param iphot: number of microcycles (inside REMC loop) type iphot: integer param constraints_force: Slope of constraints force potential type constraints_force: float Parameters: dynamics – Use of special CABS version for dynamics pathway studies :type dynamics: boolean
- rng_seed = None¶
seed for random generator
- savePdbModel(model_idx, filename='')¶
Save trajectory model into pdb file
Parameters: - model_idx – index of model in the CABS trajectory
- filename – name of the output file. If empty, it saves to model_index.pdb
- sgToPdb(output_filename='TRASG.pdb')¶
Convert TRASG (sidechains pseudoatoms) into multimodel pdb. Default filename TRASG.pdb
- trafToPdb(output_filename='TRAF.pdb')¶
Convert TRAF CABS pseudotrajectory file format into multimodel pdb (default filename TRAF.pdb)
- class pycabs.Calculate(output)¶
Inherit if you want to process data used with Monitor class.
Parameters: output (array/list) – output array with calculated results - processTrajectory(data)¶
Use it in calculate method if you parsing TRAF file, and want to calculate something on structure
Returns: array of model coordinates
- exception pycabs.Errors(value)¶
Simple error messages
- class pycabs.Info(text)¶
Simple message system
- class pycabs.Monitor(filename, calculate)¶
Class for monitoring of CABS output data. You can run it and dynamically update output arrays with calculated results.
Parameters: calculate (Calculate) – what to do with gathered data ? - daemon = None¶
if True, it will terminate when script terminates
- run()¶
Run monitor in background
- terminate()¶
Terminate monitor
- class pycabs.Template(filename)¶
Class used for storage of templates atom positions and distance calculation
Parameters: filename – path to file with template (in PDB format) Returns: Nx3 list of coordinates - distance(idx_i, idx_j)¶
Parameters: - idx_i (integer) – residue index (as in target sequence numbering)
- idx_j (integer) – residue index (as in target sequence numbering)
Returns: euclidean distance between Cα(i) and Cα(j)
- pycabs.contact_map(trajectory, contact_cutoff)¶
Compute fraction of contacts in a trajectory, where trajectory is 2D list of coordinates (trajectory[2][5] is the z-th coordinate of second atom of third model)
Parameters: - trajectory – 2D trajectory of atoms (Cα, sidegroups center of mass, etc.)
- contact_cutoff – cutoff defining contact
Returns: 2D array of fraction of contacts (number of contacts divided by trajectory length) for each pair of residue.
- pycabs.heat_map(data, x_label, y_label, colormap_label, output_file='heatmap.png', cmap='Greys')¶
Save heat map using pylab
Parameters: data (float) – 2D list of values
- pycabs.loadSGCoordinates(filename)¶
Read center of mass of sidegroups from TRASG file
Parameters: filename – path to the TRASG file Returns: 2D list of sidechains coordinates
- pycabs.loadTRAFCoordinates(filename)¶
Read trajectory file into 2D list of coordinates
Returns: 2D list of trajectory coordinates ( list[1][6] is sixth coordinate of second trajectory model = z coordinate of second atom of second model)
- pycabs.parseDSSPOutput(filename)¶
Helper function for extracting sequence and secondary structure assignments from the DSSP output. Useful for dynamics studies or other where we know protein structure.
You can download DSSP files directly from PDB server: http://www.pdb.org/pdb/files/PDBID.dssp
- pycabs.parsePDBfile(pdb_filename)¶
Function for parsing of Cα coordinates from PDB file.
Parameters: pdb_filename (string) – path to PDB file Returns: 1D list of Cα coordinates (for example: list[4] is y-th coordinate of second atom)
- pycabs.parsePorterOutput(porter_output_fn)¶
Porter (protein secondary stucture prediction, http://distill.ucd.ie/porter/) output parser. Porter emailed output looks like:
IDVLLGADDGSLAFVPSEFSISPGEKIVFKNNAGFPHNIVFDEDSIPSGVDASKISMSEE CEEEECCCCCCCCEECCEEEECCCCEEEEEECCCCCEEEEECCCCCCCCCCHHHHCCCCC DLLNAKGETFEVALSNKGEYSFYCSPHQGAGMVGKVTVN CCECCCCCEEEEECCCCEEEEEECCHHHHCCCEEEEEEC
Parameters: porter_output_fn (string) – path to the porter output file Returns: tuple (sequence, secondary_structure)
- pycabs.parsePsipredOutput(psipred_output_fn)¶
Psipred (protein secondary structure prediction, http://bioinf.cs.ucl.ac.uk/psipred/) output parser. Psipred output looks like:
> head psipred.ss 1 P C 1.000 0.000 0.000 2 K C 0.665 0.000 0.459 3 A E 0.018 0.000 0.991 4 L E 0.008 0.000 0.997 5 I E 0.002 0.000 0.998 6 V E 0.003 0.000 0.999 7 Y E 0.033 0.000 0.981
Parameters: psipred_output_fn (string) – path to the psipred output file Returns: tuple (sequence, secondary_structure)
- pycabs.rmsd(reference, arr)¶
Calculate coordinate Root Mean Square Deviation between two sets of coordinates.
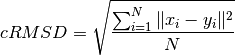
Parameters: - reference (list) – 1D list of coordinates (length of 3N)
- arr (list) – 1D list of coordinates (length of 3N)
Returns: RMSD value after optimal superimposition of two structures
- pycabs.saveMedoids(clusters, cabs)¶
Save cluster medoids in PDB file format.
Parameters: - clusters – cluster indices as a output of C Clustering Library
- clusters – list