Protein-peptide interactions play a key role in cell functions. Their structural characterization, though challenging, is important for the discovery of new drugs. The CABS-dock web server provides an interface for modeling protein-peptide interactions using a highly efficient protocol for the flexible docking of peptides to proteins [ ref 1, 2]. Some of other docking algorithms require pre-defined localization of the binding site, CABS-dock doesn’t require such knowledge. Given a protein receptor structure and a peptide sequence (and starting from random conformations and positions of the peptide), CABS-dock performs simulation search for the binding site allowing for full flexibility of the peptide and small fluctuations of the receptor backbone (see Figure 1, Movie 1, Movie 2, Movie 3 below). Additionally, the advanced option “Mark flexible regions” enable to assign a full flexibility to a selected fragment of a protein receptor structure. This feature has been used in the study of large-scale conformational changes of MDM2 receptor to p53 peptide [ ref 3] (see Movie 3). Moreover, simulation contact maps, a new CABS-dock feature, have been described in the book chapter [ ref 4].
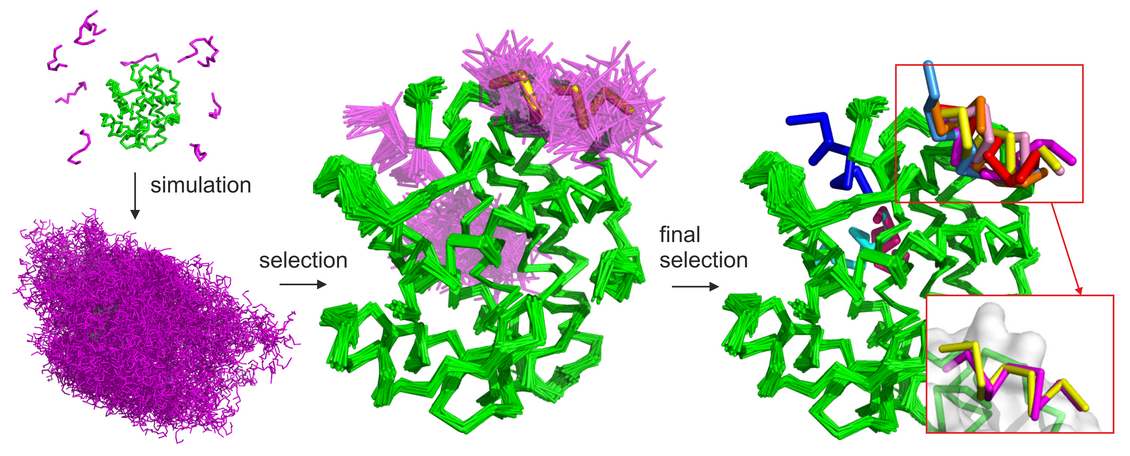
Figure 1. Basic stages of the CABS-dock protocol are illustrated on the example benchmark case (PDB ID: 2P1T). The protein receptor is colored in green, modeled peptide conformations in magenta and the reference experimental peptide structure in yellow. The following CABS-dock stages are visualized: (1) simulation start (from random conformations and positions of the peptide); (2) simulation result (10 trajectories comprising a set of 10,000 models); (3) filtering and clustering result (a set of models grouped in similar binding modes and similar peptide conformations); (4) final models (a set of 10 representative models). In the presented benchmark case, 7 of the 10 final models were docked in the native binding site (marked in red rectangle). Among these, the best accuracy model was within 1.37 Å to the native (shown in the right bottom corner superimposed on the native peptide structure).
Movie 1. CABS-dock docking simulation of, the assembly of major histocompatibility complex (MHC)-peptide structure (PDB accession code 3BWA). Experimental peptide structure is shown in green, while the simulated peptide in red. The movie shows 1 of 10 trajectories generated in a standard CABS-dock simulation run. The root mean square deviation of the peptide at the simulation end is within 1.83 Angstroms from the experimental structure.
Movie 2. CABS-dock docking simulation of the assembly of HIV-1 protease and NC-p1 peptide. During blind search for a binding site, the peptide spontaneously inserts into the receptor cavity. The lowest RMSD model is below 1 Angstrom. The movie presents three panels showing: 1 replica of the simulated peptide - in purple - and the reference experimental structure - in green (left panel); 10 replicas of the simulated peptide in a single CABS-dock simulation run (upper right panel); RMSD of the simulated peptide versus the CABS-dock energy (lower left panel).
Movie 3. CABS-dock docking simulation of binding of MDM2 protein receptor and p53 peptide with large-scale conformational changes of disordered MDM2 fragments ref 3].
The CABS-dock simulations are carried out using a CABS coarse-grained protein model. The CABS design and applications have been recently reviewed (see [ ref 5] and Movie 4).
Movie 4. Short slide presentation of the review paper on coarse-grained protein models and their applications [ ref 5.
The CABS-dock method was developed, optimized and validated during the following simulation experiments:
Movie 5. Example simulation of folding and binding of an disordered peptide (from [ ref 6]) done using the CABS-dock simulation engine.
These studies showed that the method is able to predict complex arrangements close to the native structure. Importantly, in all the validation tests mentioned above, peptides were allowed to be fully flexible and no information about the binding site or peptide conformation was used. The CABS-dock protocol consists of the following steps (see also Figure 1 and Figure 2):
Generating random structures. Random structures of the peptide are generated and randomly placed on the surface of the sphere centered at the receptor’s geometrical center (the radius of the sphere is the receptor dimension in the longest direction + 20 angstroms).
Simulation of binding and docking. The CABS-dock procedure utilizes Replica Exchange Monte Carlo dynamics with 10 replicas uniformly spread on the temperature scale. Additionally the temperatures of the replicas constantly decrease as the simulation proceeds to end on the bottom of the energy minima. On output the procedure produces 10 trajectories (one for each replica), each consisting of 1000 time-stamped simulation snapshots for a combined total of 10,000 models. During the simulation, the receptor molecule is kept in near-native conformations by a set of distance restraints binding pairs of C-alpha atoms. The restraints are selected from the distance map calculated on the input structure based on the following conditions: only C-alpha atoms located within a 5-15 Å range from each other are restrained; the minimum sequence gap between restrained residues is set to 5; violation of the restraint by less than 1 Å is not penalized; beyond that the energetic penalty increases linearly. If the user marks some of the residues as semi-flexible or fully flexible, the slope of the penalty is halved or set to 0, respectively, for all restraints assigned to the marked residues.
Selection of the final representative models is a two-step procedure:
Initial filtering. From each of the 10 trajectories, all unbound states are excluded and next 100 lowest binding energy models are selected (or less if a trajectory contains less than 100 bound states, which is rarely the case), for the next step of the procedure.
K-medoids clustering. Selected models (1000 in total) are clustered together in the k-medoids procedure. Clustering is performed 100 times with different initial medoids and k=10. Ten consensus medoids are selected as the final models.
Reconstruction of the final models. Final models are reconstructed from the C-alpha trace to an all-atom representation and subsequently undergo optimization process.
