CABS-fold server for protein structure prediction
CABS-fold description
The CABS-fold web server provides the tools for protein structure prediction from sequence only (de novo modeling) or also with the use of alternative templates (consensus modeling). The web server is based on the CABS modeling procedures ranked in previous CASP competitions ( Critical Assessment of techniques for protein Structure Prediction) as one of the leading approaches for de novo and template-based modeling. Except from template data, fragmentary distance restraints can be also incorporated into modeling process. The web server output is a coarse-grained trajectory of generated conformations, its Jmol representation and predicted models in all-atom resolution (together with accompanying analysis).
Detailed description of the CABS model: Koliński A. Protein modeling and structure prediction with a reduced representation. Acta Biochim Pol. 2004;51(2):349-71.
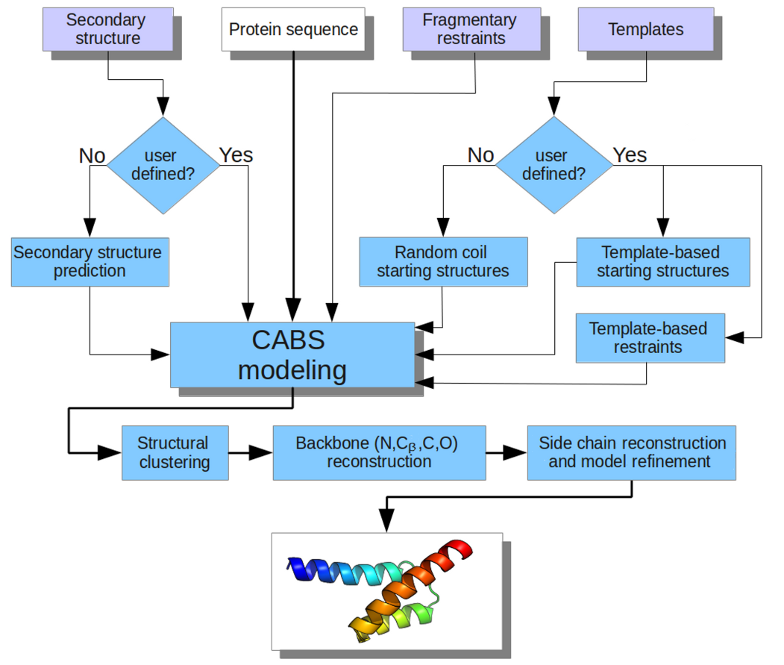
Server flowchart
The method validation
The consensus modeling protocol applied in the CABS-fold was initially tested and optimized on the set of template-based targets from CASP8. Final tests were conducted in a blind CASP9 prediction experiment by ‘LTB’ group ( group no. 400). The purpose of the LTB group was to test the consensus modeling protocol on all of CASP9 targets (thus, on all difficulty levels), using prediction results from the chosen (potentially most reliable) automated servers as templates. The comparison of the quality of the obtained models and templates showed that in the great majority of cases GDT_TS of the model was higher than the mean GDT_TS of the input structures. For five domains (T0586-D1, T0628-D1, T0540-D1, T0594-D1, T0622-D1) models provided by LTB were the best among all predictions submitted to the CASP as the first models (only one group, PRMLS, no. 065, was able to provide more, six, of the best models). These most successful prediction results were generated from mostly consistent templates, for which the method was able to produce better prediction results than any of the templates used.
The performance of the CABS model in de novo loop modeling (in practice, this is the case when a single template with missing fragments is provided in consensus modeling mode) has been validated and compared with two classical modeling tools [ ref 1]: MODELLER [ ref 2] and ROSETTA [ ref 3]. Loops of various lengths, from 4 to 25 residues, were modeled assuming an ideal target-template alignment of the remaining portions of the protein. It has been shown that classical modeling with MODELLER was on average better for short loops, while the CABS modeling was more effective for longer missing fragments.
In application to de novo modeling of large protein fragments or entire proteins (with or without fuzzy restraints) the CABS model was validated during CASP competitions as one of the leading approaches [ ref 4-6], as well as in purely de novo protein dynamics studies [ ref 7-8] demonstrating the algorithm ability to consistently fold small proteins from a highly denatured ensemble to a native-like ensemble (around 2 Å from the native), in a short CPU time.
References
- Jamroz, M. and Kolinski, A. (2010) Modeling of loops in proteins: a multi-method approach. BMC structural biology, 10(1):5
- Sali, A. and Blundell, T.L. (1993) Comparative protein modelling by satisfaction of spatial restraints. Journal of molecular biology, 234, 779-815
- Rohl, C.A., Strauss, C.E.M., Misura, K.M.S., Baker, D. (2004) Protein structure prediction using Rosetta. Method Enzymol, 383, 66–93
- Kolinski, A. and Bujnicki, J.M. (2005) Generalized protein structure prediction based on combination of fold-recognition with de novo folding and evaluation of models. Proteins, 61 Suppl 7, 84-90.
- Skolnick, J., Zhang, Y., Arakaki, A.K., Kolinski, A., Boniecki, M., Szilagyi, A. and Kihara, D. (2003) TOUCHSTONE: a unified approach to protein structure prediction. Proteins, 53 Suppl 6, 469-479.
- Debe, D.A., Danzer, J.F., Goddard, W.A. and Poleksic, A. (2006) STRUCTFAST: protein sequence remote homology detection and alignment using novel dynamic programming and profile-profile scoring. Proteins, 64, 960-967.
- Kmiecik, S. and Kolinski, A. (2008) Folding pathway of the B1 domain of protein G explored by multiscale modeling. Biophys J, 94, 726-736.
- Kmiecik, S. and Kolinski, A. (2011) Simulation of chaperonin effect on protein folding: a shift from nucleation-condensation to framework mechanism. Journal of the American Chemical Society, 133, 10283-10289.