Submitting new job and interpreting the status of processing
1. Submitting new job1.1 Consensus modeling mode (with the use of templates)
1.2 De novo modeling mode (template free)
1.3 "Advanced options" tab
1.3.1 Defining constraints
1.3.2 Adjusting simulation temperature
2. Interpreting job status
1. Submitting new job
1.1 Consensus modeling mode (with the use of templates)
See below an example screenshot of the “Submit new job” form:
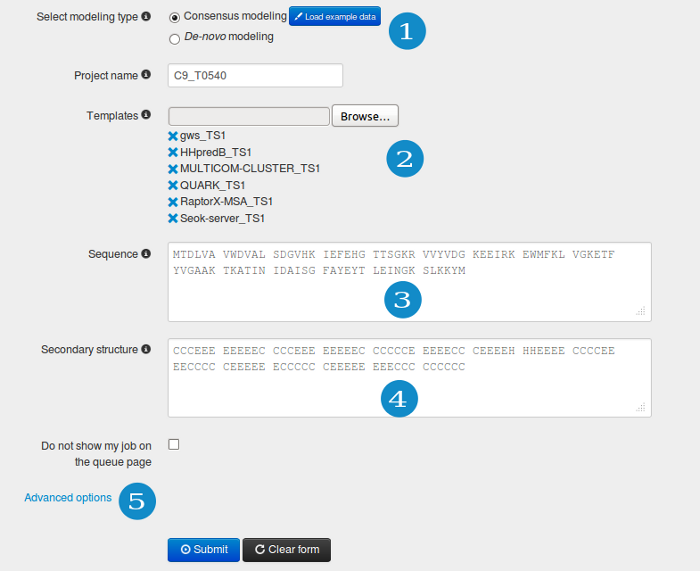
Make sure that the consensus modeling mode is chosen (1).
In this mode, at least one or more template structures should be provided in PDB format (or zip compressed PDB files) (2). Only coordinates of the Cα atoms are required, however the server also accepts data containing backbone and/or side-chains atoms. The numbering of the residues should correspond to the query sequence order. Thus the template(s), have to be already aligned to the query sequence.
Protein sequence should be entered (3) in plain text or the FASTA format. Additionally, you can provide information about the predicted secondary structure (4) (if not, the PSI-PRED method is automatically used). The secondary structure should be defined for each residue in a three letter code: H, a helix; E, an extended state (beta sheet); and C, a coil (less regular structures).
In order to add (or remove) some distance restraints click the “Advanced options” link (5), see "Advanced options" section for details.
1.2 De novo modeling mode (template free)
See below an example screenshot of the “Submit new job” form:
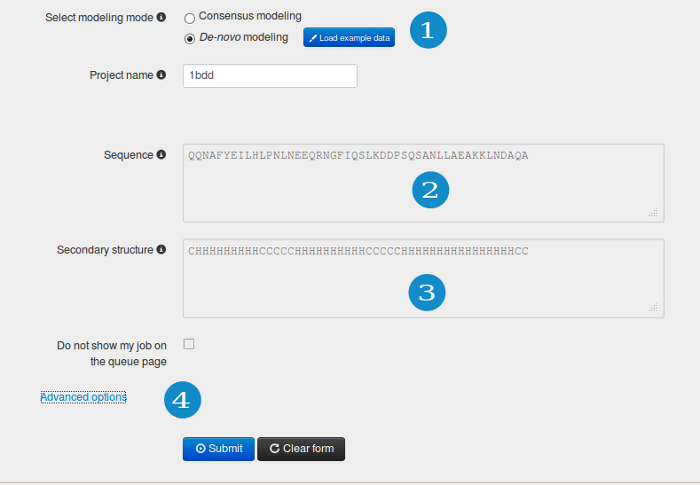
Select "de novo modeling" mode (1) to submit the prediction job based on a sequence only. The sequence should be entered (3) in plain text or the FASTA format. Additionally, you can provide information about the predicted secondary structure (4) (if not, the PSI-PRED method is automatically used). The secondary structure should be defined for each residue in a three letter code: H, a helix; E, an extended state (beta sheet); and C, a coil (less regular structures).
In “Advanced options” (4), simulation temperature can be adjusted and the distance restraints can be defined (e.g. from sparse experimental data; see "Advanced options" section for details).
1.3 "Advanced options" tab
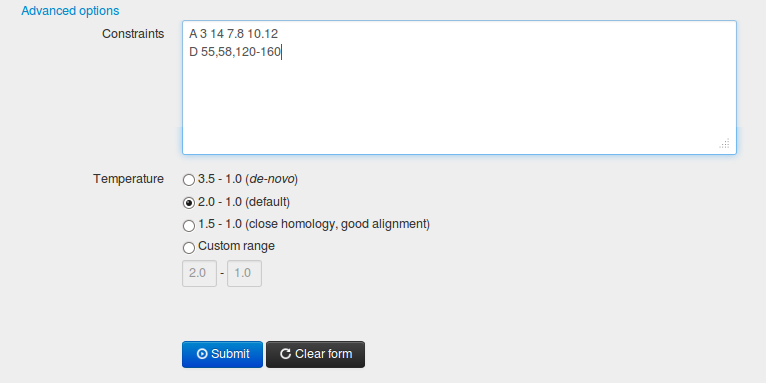
1.3.1 Defining constraints
Constraints text field allows for removing (consensus modeling mode) and/or adding (both modes) distance restraints. For example, entering a string "D 55,58,120-160" causes the deletion of all restraints (automatically generated from templates) concerning residues 55, 58 and the residues from the range 120-160. It is also possible to add new restraints, e.g. a string "A 3 14 7.8 10.12" defines the distance constraint between the 3rd and the 14th residue as the range between 7.8 and 10.12 Angstroms.
1.3.2 Adjusting simulation temperature
The “Advanced options” also allow for setting up the temperature range (in the CABS simulation algorithm (the temperature is the parameter that controls the acceptance ratio for new conformations using Replica Exchange Monte Carlo scheme). When accessing "Advanced options" the default temperature ranges can be seen. For instance, the recommended range for de novo modeling is 3.5 - 1.0, for consensus modeling 2.0 - 1.0. The temperature range can be freely modified.
In the temperature 3.5, most of the energetically unfavourable conformational changes are accepted, thus the protein chain changes its conformation very quickly adopting mostly the random coil type of a structure, while in the temperature 1.0 the chain is nearly frozen. Therefore, lowering the first temperature value, limits the possibilities of structural rearrangements with respect to the starting structure. Note that setting up the flat range of the temperatures e.g. 2.0 - 2.0 allows for the investigation of transient (less stable) folding conformers from the resulted trajectory, as shown in our protein mechanisms studies.
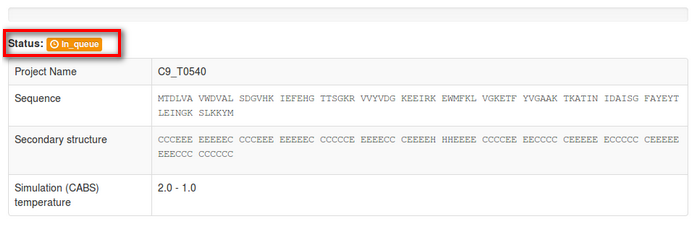
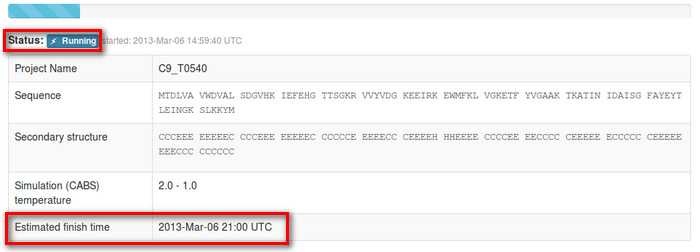
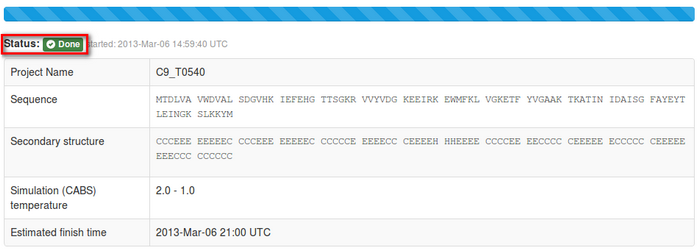
2. Interpreting job status
After clicking the Submit button, the following info will appear, which contain the unique link to your job:
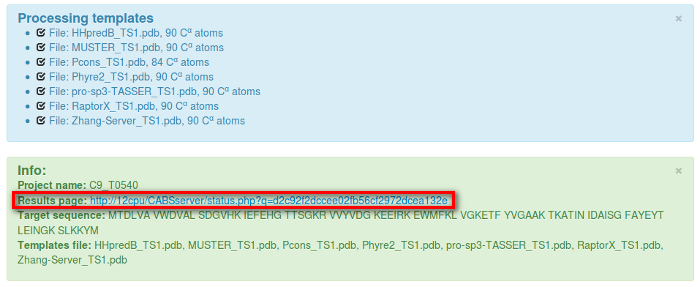
If you have chosen “Do not show my job on the queue page” it is important to save the link to your job, otherwise your job will be accessible from the queue page: ( QUEUE).
Under the unique link to your job you’ll find the job status updates and finally the job results.
The job status information will start from the “in queue status”:
The information how to access and interpret results is available here