Example results
1. De novo modeling1.1. De novo modeling without using any distance restraints data
1.2. De novo modeling with the use of sparse restraints
2. Consensus modeling
2.1. Consensus modeling - case of single template with missing fragment
2.2. Consensus modeling - case of multiple templates
3. References
1. De novo modeling
1.1. De novo modeling without using any distance restraints data
For de novo modeling without using any distance restraints data, we recommend sequence input length no longer than 120 residues. Due to enormous difficulty of the problem, the longer sequence input is very unlikely to end up with a prediction result close to the near-native structure.
- PDB code: 1BDD, length: 46 residues, RMSD of Model 1 (representative model of the most dense cluster) to the native = 2.95 Angstroms
- PDB code: 1FVQ, length: 72 residues, RMSD of Model 2 (representative model of the second most dense cluster) to the native = 3.42 Angstroms
- PDB code: 1K40, length: 126 residues, RMSD of Model 1 (representative model of the most dense cluster) to the native = 3.16 Angstroms



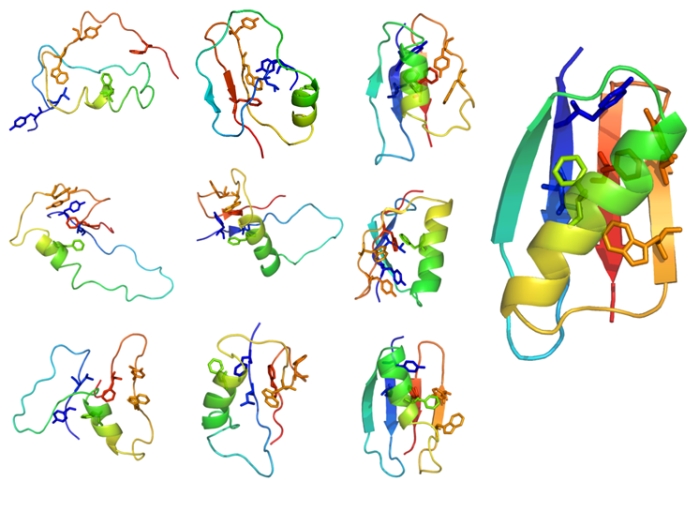
Note that setting up the flat range of the temperatures e.g. 2.0 - 2.0 allows for the investigation of transient (less stable) folding conformers from the resulted trajectory, as shown in numerous protein mechanisms studies (for example in the investigation of B1 domain of protein G [ ref 1], see example transient structures of B1 domain of protein G in the figure below).
1.2. De novo modeling with the use of sparse restraints
Below is presented the example prediction procedure with the use of the CABS de novo modeling and fuzzy restraints for the target T0201 (modeled during the CASP6 competition), as described in [ ref 2]):
The fold-recognition models were at best moderately scored by VERIFY3D, had mutually inconsistent alignments, and exhibited large differences from each other. Hence, we decided to derive additional restraints from de novo models submitted to CASP6 by ROBETTA, which had five strands, grouped together in one b-sheet with three strands and a separate b-hairpin formed by the N-terminus. All our models had a similar topology, either a ferredoxin fold or its variant with an additional N terminal b-strand added at the edge of the b-sheet and hydrogen-bonded to the C-terminal strand in an antiparallel manner. Analysis of the native structure revealed that TM1457 has a new architecture, indeed, similar to the ferredoxin fold, but with the N-terminal strand inserted into the b-sheet between the second strand and the C-terminal strand. Thus, the N- and C-terminal strands in our model are flipped compared to the native structure.
However, amazingly, the CABS refolding based on fuzzy restraints has led to a rearrangement of the two layers (helices and strands), leading to a very good superposition of the model with the native structure (Fig. 2, T0201). According to the assessment, our model_4 seems to have the best Global Distance Total Test Score (GDT_TS; 61.17) among all models submitted for this target in the course of CASP6 (our model_1 is slightly worse, but is second best according to the GDT_TS, 51.06). It is noteworthy that besides the geometry, the alignment is also very good (total C-alpha root-mean-square deviation (RMSD): 3.54 Å, without the swapped strands: 2.81 Å), which shows that our procedure can identify a reasonable sequence–structure fit for the correctly modeled core even if the initial alignment is poorly defined, and even if the peripheral elements have incorrect structure.”
Below is shown the fragment of the Figure from [ ref 2]. The figure shows cartoon diagrams of the T0201 target analyzed above (best model, the native structure, their superposition, and the possibly best template we could identify in the PDB: 1lxj). Proteins are colored according to the sequence index (N-terminus, blue; C-terminus, red), only in the superposition, where the model is in red and the native structure is in blue. Dotted elipses indicate correctly predicted (sub)structures according to global superposition (yellow) and regions with correctly predicted local structures, but incorrectly placed with respect to the remaining part of the protein (model, red; native, blue).

2. Consensus modeling
2.1. Consensus modeling - case of single template with missing fragment
Below is presented the example prediction result obtained during the
CASP9 competition for T0622 target (by 'LTB' group with the use of the
CABS-fold methodology for consensus modeling).
For the T0622 target, starting model and additional hints had been
provided by the CASP9 organizers:

source: http://predictioncenter.org/casp9/gdtplot.cgi?group=236&models=first&target=T0622-D1
In respect to the starting model, the following improvement was made:
GDT_TS - starting model: 66.80%; predicted model: 70.29%
RMSD - starting model: 7.474 Å; predicted model: 4.147 Å
2.1. Consensus modeling - case of multiple templates
Below is presented the example prediction result obtained during the
CASP9 competition for T0540 target (by 'LTB' group with the use of the
CABS-fold methodology for consensus modeling). In this case, predicted
models for T0540 target from automated structure prediction servers
were used as templates. The following models/templates were used (the
model name denotes the server name): HHpredB_TS1, Pcons_TS1,
pro-sp3-TASSER_TS1, MUSTER_TS1, Phyre2_TS1, RaptorX_TS1,
Zhang-Server_TS1
Figure below shows superimposition of the CA traces of the templates, native structure and predicted model.

The predicted model was significantly better than any of the templates:
GDT_TS - mean for the templates: 59.93%; the best template: 62.78%; predicted model: 68.61%
RMSD - mean for the templates: 4.737 Å; the best template: 4.259 Å; predicted model: 3.570 Å
According to the the official CASP assessment the predicted model was ranked as the most accurate among all predictions submitted as first model (see plot below).

Figure: GDT analysis: largest set of CA atoms (percent of the modeled structure) that can fit under DISTANCE cutoff: 0.5A, 1.0A, 1.5A, ... , 10.0A.
source: http://predictioncenter.org/casp9/gdtplot.cgi?group=236&models=first&target=T0540-D1
3. References
- Kmiecik, S. and Kolinski, A. (2008) Folding pathway of the B1 domain of protein G explored by multiscale modeling. Biophys J, 94, 726-736. doi: 10.1529/biophysj.107.116095
- Kolinski, A. and Bujnicki, J.M. (2005) Generalized protein structure prediction based on combination of fold-recognition with de novo folding and evaluation of models. Proteins, 61 Suppl 7, 84-90. doi:10.1002/prot.20723