CABS-flex server for fast simulation of protein structure fluctuations
CABS-flex is an efficient procedure for the simulation of structure flexibility of folded globular proteins. It is based on the CABS model, the well-established coarse-grained protein modeling tool. The CABS may generate consistent protein dynamics at highly reduced (3 orders of magnitude) cost, although with some decrease of resolution. The CABS-flex server follows the work of Jamroz, Orozco, Kolinski and Kmiecik [ ref 1], where the authors demonstrated, that the consensus view of protein near-native dynamics obtained from 10-nanosecond MD simulations (all-atom, explicit water, for all protein metafolds using the four most popular force-fields), is consistent with the CABS dynamics. This observation have been confirmed in further extensive validation tests [ read NAR paper].
The only data required as an input is a protein structure in PDB format (or a protein PDB code). The input structure is used as a starting point for the CABS simulation of near-native dynamics. The resulting trajectory is automatically analyzed and processed to provide the useful description of protein dynamics in the following outputs:
- a set of protein models (in an all-atom resolution) reflecting the most dominant structural fluctuations in the near-native ensemble,
- fluctuation profiles: residue fluctuation profile (showing the relative propensity of protein residues to deviate from an average dynamics structure), and RMSD profiles (displayed for each representative model, showing the deviation of protein model residues from the input structure).
An important attribute of protein models generated by the CABS is that their spatial resolution (in C-alpha chain format) allows for reconstruction of all-atom representation of physically realistic models. The output set of all-atom models is generated through trajectory clustering (by k-means method) and subsequent multi-step reconstruction and optimization procedures (the procedures were extensively tested in previous protein dynamics and structure prediction studies).
The CABS-flex server pipeline is presented below (different kinds of output data are marked in blue):
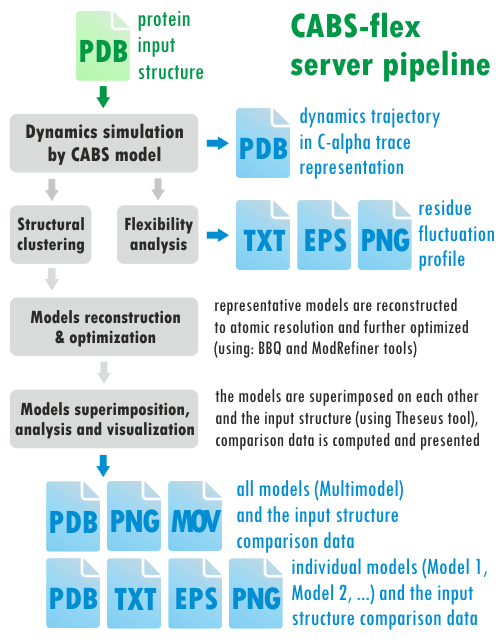
The CABS-flex server employs the following tools:
- CABS simulation model [ ref 2]
- BBQ method for the reconstruction of main chain atom coordinates from alpha carbon trace [ ref 3]
- ModRefiner for the reconstruction and optimization of predicted protein models [ ref 4]
- Theseus software package for the superimposition of multiple protein models [ ref 5]
- Gnuplot software package for creating online presented plot pictures and eps plot files [ ref 6]
- Pymol visualization software for creating online presented pictures of superimposed models [ ref 7]
Note that by using the predicted models from the CABS-flex server you agree with all the licenses of external tools listed above.
Benchmarks
Comparison of CABS Dynamics with Molecular Dynamics on 393 protein data set.
References
- Jamroz M., Orozco M., Kolinski A., Kmiecik S. 2013, Consistent View of Protein Fluctuations from All-Atom Molecular Dynamics and Coarse-Grained Dynamics with Knowledge-Based Force-Field, J. Chem. Theory Comput., 9 (1), 119–125 doi: 10.1021/ct300854w
- Kmiecik S., Koliński A. (2007), Characterization of Protein Folding Pathways by Reduced-space Modeling. Proc. Natl. Acad. Sci. USA, 104(30):12330–5
- Gront D., Kmiecik S. and Koliński A. (2007), Backbone building from quadrilaterals: A fast and accurate algorithm for protein backbone reconstruction from alpha carbon coordinates. J. Comput. Chem., 28: 1593–1597. doi: 10.1002/jcc.20624
- Dong Xu and Yang Zhang (2011). Improving the Physical Realism and Structural Accuracy of Protein Models by a Two-step Atomic-level Energy Minimization. Biophysical Journal, vol 101, 2525-2534
- Theobald, D. L., Wuttke D. S. (2006). THESEUS: Maximum Likelihood Superpositioning and Analysis of Macromolecular Structures. Bioinformatics 22 (17): 2171–2. doi:10.1093/bioinformatics/btl332.
- Williams T., Kelley C. (2011). Gnuplot 4.5: an interactive plotting program
- The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC
Server statistics
| Current server load | 0.2/12 |
| Uptime | 42w 3d 1h 33m 1s |
| Projects, finished | 1699 |
| Projects, pending | 0 |
| Projects, total | 1714 |
| Modelled aminoacids, total | 257799 |
© Laboratory of Theory of Biopolymers, Faculty of Chemistry, University of Warsaw 2013